
The Lungs and Lupus
This site is intended for healthcare professionals as a useful source of information on the diagnosis, treatment and support of patients with lupus and related connective tissue diseases.
KEY POINTS:
• Respiratory disease is common in SLE.• Respiratory disease in SLE can affect chest wall, pleura, airways, lung parenchyma and pulmonary vasculature.
• Symptoms are similar to other serious pathologies e.g. PE or pneumonia.
Introduction
Chest disease commonly complicates SLE and may sometimes be the presenting or dominant disease manifestation. The types of chest and lung diseases are associated with SLE are summarised in Table 1. Patients with SLE are also more susceptible to lower respiratory tract infections as a result of the use of immunosuppressive drugs. Finally, some medications used to treat SLE can, themselves, cause lung disease.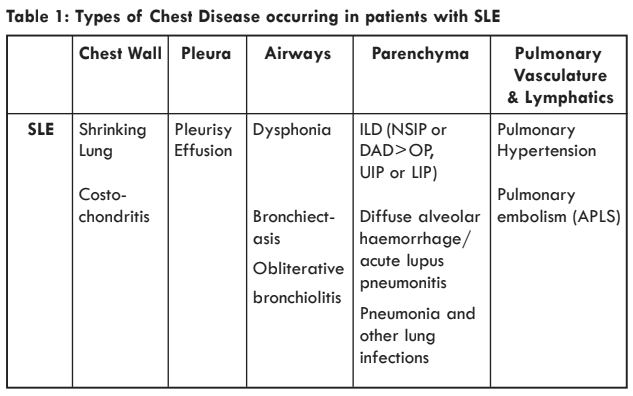
SLE: systemic lupus erythematosus; ILD: interstitial lung disease; NSIP: non specific interstitial lung disease; DAD: diffuse alveolar damage; OP: organising pneumonia; UIP: usual interstitial pneumonia; LIP: lymphocytic interstitial pneumonia; APLS: antiphospholipid antibody syndrome.
Chest wall disease
Shrinking Lung Syndrome (SLS) is rare (0.5-1% of patients with SLE). The pathophysiology of this disease is still poorly understood and possibly relates to respiratory muscle weakness (predominantly the diaphragm), caused by an unknown myopathic process without documented abnormality of other lung tissue. There is, however, insufficient evidence to support this. SLS should be considered when lupus patients present with unexplained dyspnoea or chest pain in the presence of reduced lung volumes. Interstitial lung disease should be absent or insufficient to explain the loss of lung volume. SLS is a diagnosis of exclusion. SLS usually responds to corticosteroids and immunosuppressants but a chronic restrictive ventilatory defect may persist.Costochondritis is inflammation affecting the costochondral joints of the chest wall. This occurs commonly in SLE and presents as chest pain (anterior, posterior, or both), which is worse on deep inspiration. The chest wall is tender over the relevant joints and pressure at these locations reproduces the pain. Costochondritis may be relieved by topical NSAIDs, simple analgesics, oral NSAIDs, or, if these approaches fail, by a course of corticosteroids. Be aware that rib pain in a patient with SLE and osteoporosis (or a history of chronic corticosteroid use) may represent a rib or vertebral fracture, rather than costochondritis. A CXR may be normal within the first few days after the fracture but, after a week, callus should be evident.
Pleural disease
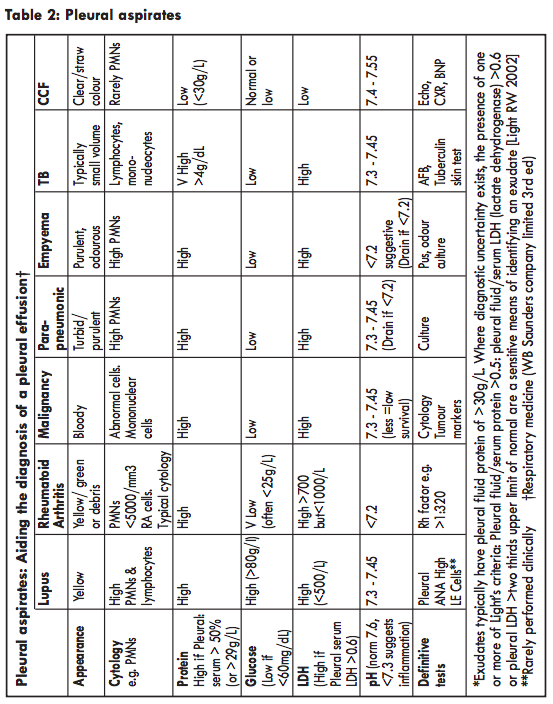
Up to 60% of patients with lupus experience pleuritic chest pain at some point in the disease course. In some cases, this can be associated with a serous/serosanguinous pleural effusion, typically exudative with a high protein and lactate dehydrogenase (LDH) versus serum (LDH here is, however, lower than in Rheumatoid Arthritis and glucose is higher). Table 2.Given that chest signs and symptoms can be relatively common, the decision to investigate will depend on the severity of symptoms and whether they present alongside other features that arouse suspicion of new/significant intrathoracic disease. For example, diagnostic aspiration of a new effusion in the presence of elevated CRP is likely to be useful, more so than in an individual with recurrent lupus related pleurisy which has responded well to anti-inflammatory medications. Pleural biopsy is less useful, being poorly specific and, therefore, rarely performed. It should also be remembered that the presence of lupus does not preclude the presence of thoracic disease of other aetiologies.
Lupus associated pleuritis responds to NSAIDs in mild cases or, where severe, may require increased dose corticosteroids. Anti-malarials are useful for refractory cases. Colchicine may also be useful in acute pleuritis.
Airways
Patients with lupus with or without Sjögren's Syndrome may develop dysphonia, i.e. difficulty in speaking. Common causes include an extension of the Sjögren's-related xerostomia to the oropharynx, post-nasal drip, use of oral corticosteroids (e.g. for recurrent mouth ulcers), or use of inhaled corticosteroids for bronchospasm. However, rarer causes should be considered in the appropriate context. Recurrent laryngeal neuropathy as a manifestation of mononeuritis and laryngomalacia resulting from relapsing polychondritis can sometimes occur as part of the lupus spectrum. Important non-lupus pathologies, including laryngeal nodules or malignancy, should also be excluded.A new presentation of hoarseness should prompt a referral to ENT, since malignancy should be excluded. However, more common causes are inhaled cortisteroids, post-nasal drip, xerostomia or hypothyroidism.
Examination should include the thyroid for the presence of a goitre and cervical lymph nodes, Schirmer’s test, general examination for other signs of small vessel vasculitis (e.g. rashes, other neuropathies, active renal sediment) or relapsing polychondritis (polyarthralgia, costochondritis, aortic regurgitation).
Investigations include those required to diagnose Sjögren's Syndrome (autoimmune serology, minor salivary gland biopsy), vasculitis screen, neck ultrasound and, in some circumstances, referral to ENT for nasendoscopy/laryngoscopy.
Treatment of xerostomia includes assiduous dental hygiene, use of high fluoride toothpaste, rinsing out mouth after use of corticosteroid bronchodilators, use of oral moisturisers (e.g. artificial saliva or oral gel preparations). Post-nasal drip may respond to treatment of sinusitis or, sometimes, to short-term use of an inhaled nasal steroid.
Bronchiectasis is dilatation and thickening of airways proximal to the terminal bronchioles. This usually results from chronic airway inflammation. It occurs in ~ 20% of patients with SLE, most commonly in those with secondary Sjögren's syndrome. Bronchiectasis should be suspected in patients with a persistent, productive cough or in patients with recurrent lower respiratory tract infections. It is evident on a high resolution chest CT (HRCT). Treatment includes respiratory physiotherapy with postural drainages, mucolytic agents, e.g. carbocysteine, and prompt culture of sputum during exacerbations, with selection of antibiotics based on culture sensitivities, where possible.
Obliterative bronchiolitis (OB) is rare in SLE. It reflects obstruction of the small airways by surrounding fibrosis. The aetiology is unclear. Radiologically, OB is characterised by a “mosaic attenuation pattern” on HRCT and this is seen more easily in expiration images. OB responds poorly to corticosteroids, but macrolide antibotics (e.g clarithromycin) and immunosuppressives may be helpful. In cases where OB is progressive and resistant to treatment, a lung transplant may be considered.
Lung Parenchyma
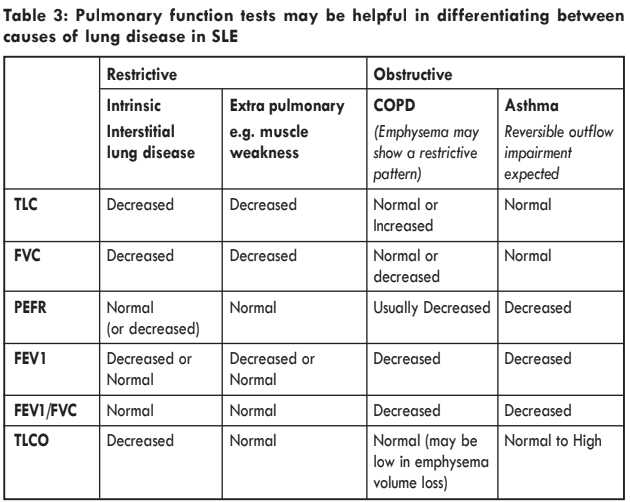
Interstitial lung disease (ILD) is well recognised in connective tissue disorders. Symptoms include progressive dyspnoea and non-productive cough.The chest X-ray shows coarse reticulonodular densities in the lower zones but, as with lung function tests, cannot easily differentiate between an acute alveolitis and a chronic picture of fibrosis; this requires serial investigations. Lung function tests generally show a proportionate reduction in both lung volumes (e.g. FVC) and gas transfer (DLCO). High-resolution CT images can detect progressive parenchymal change. ‘Ground glass’, focal alveolar opacities, imply active inflammation (most commonly NSIP), potentially treatable with steroids, whereas macroscopic, honeycomb-like
cysts are more consistent with end-stage irreversible fibrosis (UIP). ILD associated with connective tissue disease may be indistinguishable from idiopathic fibrosis, but the CTD-associated disease is generally more responsive to steroids and immunosuppressive therapy.
Organising Pneumonia with bronchiolitis obliterans (BOOP) often presents with more sub-acute symptoms of cough, dyspnoea, fever and constitutional un wellness. Middle-aged women are most typically affected.
Airflow limitation arises from submucosal oedema with peribronchiolar inflammation and fibrosis, primarily involving respiratory bronchioles in the absence of diffuse parenchymal disease, thought to arise from plugs of fibrous tissue in bronchioles and alveolar ducts. Prognosis is better than that for those with a standard interstitial pneumonia (NSIP), particularly when BOOP is the diagnosis since there is often an excellent response to corticosteroids.
Whilst chest radiographs in BOOP may show focal alveolar infiltrates with striking air bronchograms, there are often no changes in pure BO where the lung parenchyma is spared. Pulmonary function tests show a restrictive defect. Clinical and serological markers may also correlate with such chronic lung disease in lupus. Both Scleroderma-like symptoms (e.g. Raynaud’s) and Anti-SSa (Ro) antibody seem to be more common with restrictive defects and a reduction in transfer factor. Another important differential is anti-Jo-1 associated myositis/lung disease.
All such patients should be assessed in a specialist centre.
Pulmonary haemorrhage complicating lupus is rare and can range from a mild sub-clinical chronic form to massive life threatening haemorrhage with a mortality of up to 90%. Patients may have haemoptysis at presentation, but sometimes manifestations can be clinically non-specific and include unexplained anaemia, cough and dyspnoea. Risk factors include known lupus, high levels of anti-dsDNA antibodies and active extra-pulmonary disease. Glomerulonephritis is also often present in these patients. Important differential diagnoses include anti-GBM disease and ANCA associated vasculitis (e.g. granulomatosis with polyangiitis, microscopic polyangiitis or eosinophilic granulomatosis with polyangiitis). Pulmonary function test results showing increased diffusing capacity for carbon monoxide is a sensitive test for acute alveolar haemorrhage although it can betechnically challenging to perform in severe cases. Increasingly, bloods tained samples from bronchoalveolar lavage or progressive chest radiograph in association with a fall in haemoglobin are highly suggestive. Lung biopsies show capillaritis with immunoglobulin or immune complex deposition although, clinically, these are rarely indicated. Treatment options include high dose steroids and cyclophosphamide and, in severe cases, plasma exchange, although the latter can be hazardous in the presence of thrombocytopenia or coagulation disorders.
Pulmonary Vasculature
Pulmonary hypertension is defined as elevated pulmonary artery pressure(more than 25mmHg at rest/30mmHg on exertion) and can be of primary origin (pulmonary arterial hypertension) or secondary to interstitial lung disease or pulmonary emboli. The presence of antiphospholipid antibodies in lupus predisposes to a thromboembolic aetiology. The incidence of pulmonary hypertension is also increased in the presence of Raynaud’s phenomenon. Symptoms including lethargy, fatigue, angina, syncope and dyspnoea on exertion are relatively non-specific. Diagnosis is, therefore, aided by the presence of distended jugular veins (with prominent a and v waves), right ventricular heave, fixed split S2, hepatomegaly, ascites and peripheral oedema. Investigations should include ECG (changes are consistent with right ventricular hypertrophy), CXR (cardiomegaly combined with prominent pulmonary arteries can lead to straightening of the left heart border on the background of oligaemic lung fields) and lung function tests (which show a disproportionare reduction of DLCO relative to FVC). Echocardiographic estimate of pulmonary artery pressures from tricuspid regurgitant velocity may be suggestive but formal diagnosis requires right heart catheterisation which may be accompanied by vasodilator challenge.
Treatment requires involvement of a specialist pulmonary hypertension centre. It includes control of any underlying activity of interstitial lung disease with steroids and cytotoxic agents, combined with vasodilators. Prognosis is generally poor but is improved in secondary pulmonary hypertension if an underlying cause can be identified and reversed.
Sudden onset pleuritic chest pain with acute respiratory compromise should arouse suspicion of pulmonary embolism (PE), particularly if the ECG shows sinus tachycardia or right heart strain (classically “S1Q3T3”). The incidence of thromboembolic events is increased in the presence of antiphospholipid antibodies (aPL) which strongly predispose to arterial and venous thromboses.
Lupus anticoagulants are so named because in vitro clotting is inhibited. In vivo, paradoxically, thrombotic events are more common. The APTT is unreliable, being prolonged in only half of SLE patients with lupus anticoagulant and often not at all in the presence of anticardiolipin antibodies (aCL). More reliable tests (such as ELISA for aCL and beta2 glycoprotein 1, or dRVVT for lupus anticoagulant) should be ordered early in patients with unexplained thrombotic events.
Standard risk factors e.g. smoking, oral contraceptives, malignancy, surgery and immobility may also co-exist in lupus and should be considered when establishing probability of venous thrombosis. Corticosteroids also increase the risk of venous thromboembolism, both acutely and chronically.
Patients in whom there is a high clinical suspicion of PE in the presence of pre- existing lung disease and/or serological risk factors (LAC or high titre aPL), should be anticoagulated pending further investigation (CTPA is usually preferred due to a higher specificity when there is previous/chronic lung damage that renders V:Q scanning less accurate). Long term anti-coagulation is generally recommended in patients with high titre aPL who have had a pulmonary embolus.
Lower Respiratory Tract Infections
Patients with SLE have increased susceptibility to lower respiratory tract infections. The mechanisms contributing to this susceptibility include defective macrophage antibacterial activity, complement deficiency impairing opsonisation and immune presentation of bacterial antigens, immunosuppression with corticosteroids and DMARD’s, hypogammaglobulinaemia and late-onset neutropenia following treatment with rituximab. Patient with bronchiectasis are at particular risk due to reduced clearance of respiratory secretions. Unusual and more aggressive opportunistic microorganisms e.g. Aspergillus, Cryptococcus, Pneumocystis jirovecii (carinii), CMV and Nocardia are, therefore, more likely. Hypocomplementaemic patients also have increased sensitivity to Streptococcus pneumoniae and Neisseria meningitidis, both of which may result in fatal infection.Tuberculosis (TB) is also more common in lupus and should be excluded if haemoptysis, weight loss or fever persist. TB may also give rise to isolated pleural involvement and suspicion should be high where systemic upset occurs in ‘at risk populations’ and those in contact with individuals with AFB (Acid-Fast Bacilli) positive sputum. A chest radiograph may identify multiple opacities or nodular (potentially cavitating) masses.
In any infection, accurate early diagnosis and identification of organisms and sensitivities are crucial. Whilst infection in the presence of other lupus-related lung disease can be difficult to diagnose, this may be aided by a high CRP which, in lupus, is generally not elevated, even in the presence of active inflammation. Prompt “appropriate” treatment is essential to rapid recovery, particularly in septicaemia where “Surviving Sepsis” guidelines should be followed. In hypocomplementaemic patients who suffer recurrent infection there is also an argument for prophylactic antibiotics. SLE patients with ILD or bronchiectasis, or patients in whom non-urgent B cell depletion therapy is planned, should have titres for anti-pneumococcal antibodies and anti-Haemophilus influenza checked. Patients with low titres can be vaccinated with Pneumovax or Prevenar (for immunity to Pneumococcus) or with Menitorix (for immunity to H. influenzae).
Drug-induced Lung Disease in Lupus
A number of drugs used to treat lupus have been associated with lung disease including methotrexate, cyclophosphamide and rituximab. Methotrexate-related lung toxicity is rare and presents with a picture of acute hypersensitivity pneumonitis, which usually presents early (weeks to few months) after initiating treatment with the drug. Usual clinical symptoms include acute onset breathlessness, progressive non-productive exertional cough and chest pain. This is well recognised in rheumatoid arthritis but is unusual in SLE. Indeed, methotrexate is sometimes used as a treatment for SLE-related ILD.Hypersensitivity pneumonitis can also occur with a range of medications. Lung function tests usually show a restrictive defect with a drop in transfer factor. CT chest reveals diffuse ground glass opacities, often with air-trapping. BAL is not commonly performed but drug induced lung disease may be associated with raised eosinophils in the lavage fluid. The management includes cessation of the implicated drug. Short courses/pulses of corticosteroid may also be useful. Hypersensitivity pneumonitis associated with rituximab may take months to resolve.
Dr Frances Hall
Consultant Rheumatologist
Addenbrooke’s Hospital
Cambridge CB2 2QQ
Consultant Rheumatologist
Addenbrooke’s Hospital
Cambridge CB2 2QQ
Revised and updated from 2009 edition
by Dr Richard Lee and Prof Kevin Davies
by Dr Richard Lee and Prof Kevin Davies
 ©2024 LUPUS UK (Registered charity no. 1200671)
©2024 LUPUS UK (Registered charity no. 1200671)